Learn how the IRB process works to ensure participant safety and quality research.
Takeaways:
- The institutional review board (IRB) process is intended to ensure study participant safety and high-quality research.
- Work with colleagues familiar with the process to help you navigate it.
- As you develop the timeline for your research project, include the IRB application process and schedule.
WHEN you participate in research, you have the opportunity to create new knowledge and evidence to inform patient care and nursing practice. And you’re likely to interact with an institutional review board (IRB) or work on an IRB-approved project as a principal investigator, co-investigator, sub-investigator, research assistant, or team member. Whatever your role, understanding human subject protections is an important step in the research process.
Mandates published in the Code of Federal Regulations (CFR) and enforced by the U.S. Office for Human Research Protections (OHRP) guide IRB functions, board composition, membership qualifications, and the IRB role in protecting human subjects. You’ll also want to know what kinds of projects generally require IRB review, how to complete an application, and what you may expect after submission. And don’t forget to familiarize yourself with your organization’s specific IRB protocols and procedures.
IRB responsibility
IRBs are responsible for confirming that research protocols are developed in accordance with federal regulations based on Protection of Human Subjects (Common Rule) guidelines. Respect for individuals, beneficence, and justice are the regulation’s guiding ethical principles. The 1991 Common Rule (codified in separate regulations by 15 federal departments and agencies) outlines basic IRB provisions, informed consent, and compliance assurances.
The primary purpose of an IRB is to ensure that the rights, welfare, and well-being of people participating in research activities are protected in accordance with federal regulations. This is achieved in part by delineating IRB committee composition, including the number, backgrounds, and characteristics of members.
Regulations require that at least five members from different disciplines and backgrounds review the research activities commonly conducted by the institution. For example, nurse representation is needed if nurses within the organization commonly conduct studies involving human subjects. At a minimum, a convened IRB meeting must include five voting members, and those members must be diverse on a number of attributes, including gender, race, ethnicity, and profession. In addition, most members should be scientists with sufficient expertise to judge design safeguards across several content areas. At least one member must have a non-scientific background, and at least one must be from the community and not affiliated with the organization (for example, former patients, business owners, or clergy).
What is research?
 The IRB is tasked with reviewing research studies to protect participants, but determining what qualifies as research according to the Federal Regulation standards can be difficult. According to the OHRP, research is “a systematic investigation, designed to develop or contribute to generalizable knowledge.” The intent to develop generalizable knowledge is what makes research distinct from quality improvement projects, which typically focus on making localized process improvements (for example, on a unit or in a hospital or clinic). Research that will require IRB review includes pilot studies with human subjects and studies with human subjects that use medical or other devices (apps, drugs, food, supplements). In addition, if identifiable subject information is used, an IRB review will be required. Research that doesn’t need IRB review includes activities intended only for quality improvement and data collected only for internal departmental or administrative purposes.
The IRB is tasked with reviewing research studies to protect participants, but determining what qualifies as research according to the Federal Regulation standards can be difficult. According to the OHRP, research is “a systematic investigation, designed to develop or contribute to generalizable knowledge.” The intent to develop generalizable knowledge is what makes research distinct from quality improvement projects, which typically focus on making localized process improvements (for example, on a unit or in a hospital or clinic). Research that will require IRB review includes pilot studies with human subjects and studies with human subjects that use medical or other devices (apps, drugs, food, supplements). In addition, if identifiable subject information is used, an IRB review will be required. Research that doesn’t need IRB review includes activities intended only for quality improvement and data collected only for internal departmental or administrative purposes.
Determining whether research involves human subjects also is defined by the federal guidelines and depends on many factors, including the type of interaction, the type of data collected, and the subject population. (See Protecting research subject identity.) For example, a plan for the confidentiality of the data must be evident for respondents when a survey asks about sensitive or volatile topics. Using pre-existing data with identifiers that have been collected for another purpose is likely to require IRB review and approval before use, particularly data containing personal identifiers.
Participant risk
Study applications are evaluated on the participants’ potential psychological, social, economic, physical, and legal risks and the measures that are taken to mitigate them. Federal regulations define minimal risk as “the probability and magnitude of harm or discomfort anticipated in the proposed research are not greater, in and of themselves, than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests.” Both the probability and magnitude of possible harm can vary from minimal to significant.
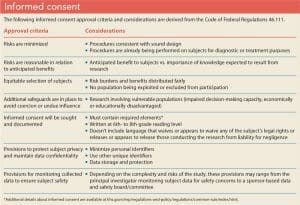 Although removing all risk from research participation is impossible, IRBs work to balance the rights of individual subjects with developing knowledge to further society as a whole. The IRB is responsible for confirming that research protocols meet federal guidelines and that they’re written in accordance with the Common Rule. IRBs seek to ensure that study designs maximize benefits while minimizing potential harm and that those who agree to participate in research do so voluntarily with a full understanding of participation expectations and risks. (See Informed consent.) Healthcare providers conducting research must describe the voluntary nature of participation and the absence of coercion (for example, excessive cash payments or expensive gifts) in the IRB application regardless of the population being recruited.
Although removing all risk from research participation is impossible, IRBs work to balance the rights of individual subjects with developing knowledge to further society as a whole. The IRB is responsible for confirming that research protocols meet federal guidelines and that they’re written in accordance with the Common Rule. IRBs seek to ensure that study designs maximize benefits while minimizing potential harm and that those who agree to participate in research do so voluntarily with a full understanding of participation expectations and risks. (See Informed consent.) Healthcare providers conducting research must describe the voluntary nature of participation and the absence of coercion (for example, excessive cash payments or expensive gifts) in the IRB application regardless of the population being recruited.
IRB process
 Before your research can begin, you’ll need to develop a timeline that includes application completion and IRB review.
Before your research can begin, you’ll need to develop a timeline that includes application completion and IRB review.
Timeline and application
As you develop a timeline for your study, familiarize yourself with your institution’s IRB application process and schedule any required additional approvals (for example, from a nurse manager or nursing shared governance council). Allow time for you and any co-investigators to complete and document human ethics training as required by your organization. Based on the data that you’re collecting, consider the level at which your project might be reviewed. If your study is scheduled for a full board review, adhere to all deadlines, especially if your timeline is short. (See IRB review categories.)
Submitting a complete and detailed application is essential to preventing unnecessary delays. Be specific, avoid using acronyms, define all terms, and detail all procedures. (See IRB application: What to expect.)
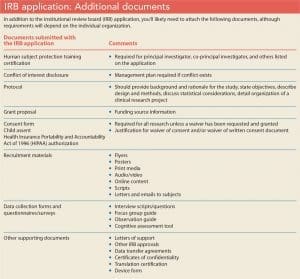
 Complete any additional sections required by your institution (if an item doesn’t apply to your study, don’t leave it blank, note that it’s not applicable) and attach any necessary documents. (See IRB application: Additional documents.) Although the timeframe from protocol submission to decision may vary depending on your organization and the study’s complexity, allow 1 to 3 months for IRB review.
Complete any additional sections required by your institution (if an item doesn’t apply to your study, don’t leave it blank, note that it’s not applicable) and attach any necessary documents. (See IRB application: Additional documents.) Although the timeframe from protocol submission to decision may vary depending on your organization and the study’s complexity, allow 1 to 3 months for IRB review.
IRB review
Typically, the review process of exempt or expedited protocols is more rapid than those requiring a full board review. Protocols requiring full board review typically pose
more than a minimal risk to subjects (for example, interventional clinical trials), involve a vulnerable population (for example, children), use deception (for example, telling subjects that they’ll engage in a cooperative task with other subjects when in fact they’re interacting with study personnel), or collect potentially sensitive information (for example, about illegal activities).
 When full board review is required, the application and supporting documents are placed on the board agenda and distributed to all committee members. A primary and secondary reviewer are assigned to examine the documents in accordance with federal guidelines. At the meeting, the reviewers present relevant study details, including the purpose, participants, and procedures. The board members discuss the risks and benefits of the study and its alignment with federal guidelines to ensure all criteria for approval have been met. Any regulatory findings are confirmed, a recommendation is made, and a final vote taken.
When full board review is required, the application and supporting documents are placed on the board agenda and distributed to all committee members. A primary and secondary reviewer are assigned to examine the documents in accordance with federal guidelines. At the meeting, the reviewers present relevant study details, including the purpose, participants, and procedures. The board members discuss the risks and benefits of the study and its alignment with federal guidelines to ensure all criteria for approval have been met. Any regulatory findings are confirmed, a recommendation is made, and a final vote taken.
The possible disposition of a study application includes approval with or without stipulations, deferral, or disapproval. Approval with conditions or stipulations requires that investigators make specified changes to the research protocols and/or informed consent documents, submit clarifications, or provide additional documents. Remember that research can’t begin until the IRB has given full unconditional approval.
Be prepared
Before you begin, seek the advice of someone who’s familiar with the IRB application process at your organization and ask him or her to proofread your application and provide feedback. If your attendance is required at a full board meeting, rehearse your presentation and request an experienced colleague accompany you to the meeting.
The IRB process may seem arduous, but it’s necessary to ensure participant safety and successful research that benefits healthcare providers, patients, and the community. (See 10 tips for IRB success.)

Amy Marzinsky is the clinical research coordinator in the division of pediatric surgery at the University of North Carolina School of Medicine – Chapel Hill (UNCCH) and a member of the Non-Biomedical Institutional Review Board (IRB) Committee E in the UNC-CH Office of Human Research Ethics. Cheryl A. Smith-Miller is a nurse scientist, nursing quality and research, at the University of North Carolina Medical Center in Chapel Hill, vice-chair of the Non-Biomedical IRB Committee E in the UNC-CH Office of Human Research Ethics, and adjunct faculty at UNC-CH school of nursing.
Selected References
American Nurses Association (ANA). 2019 New Knowledge, Innovations & Improvements: Criteria for Nursing Excellence. Washington DC: ANA; 2019.
Hung HY, Wang YW, Feng JY, Wang CJ, Lin EC, Chang YJ. Evidence-based practice curriculum development for undergraduate nursing students: The preliminary results of an action research study in Taiwan. J Nurs Res. 2019;27(4):e30.
Office for Human Research Protections. Revised Common Rule regulator text. April 2, 2019. hhs.gov/ohrp/regulations-and-policy/regulations/revised-common-rule-regulatory-text/index.html
Office for Human Research Protections. Federal policy for the protection of human subjects (‘Common Rule’). 2016. hhs.gov/ohrp/regulations-and-policy/regulations/common-rule/index.html
O’Mathúna DP. How should clinicians engage with online health information? AMA J Ethics. 2018;20(11):E1059-66.
U.S. Department of Health, Education, and Welfare. The Belmont report: Ethical principles and guidelines for the protection of human subjects of research. April 18, 1979. hhs.gov/ohrp/regulations-and-policy/belmont-report/read-the-belmont-report/index.html
U.S. Food & Drug Administration. Institutional review boards frequently asked questions. January 1998. fda.gov/RegulatoryInformation/Guidances/ucm126420.htm#IRBOrg